Using ProCARs¶
ProCARs is a Python package, which has been developed using Python 2.7.5.
Installing ProCARs¶
To run ProCARs on your dataset(s), you have two possibilities, depending on your use: installing it or running it from the sources.
For a classic use, you can install the package procars. For that, you also have two possibilities: installing it on your system as every Python package, or installing it in development mode. You can see here-below the difference between these two types of installations, and choose the one you need, according to your use.
Note
Note that you can always uninstall ProCARs if you want to install a new version instead.
Development mode¶
If you want to install the package procars while still working on modifying the scripts, or being able to download and run latest versions, just do:
sudo pip install -e .
or:
sudo python setup.py develop
Your changes will then be taken into account. As you installed the package, you will be able to run ProCARs from any directory in your computer.
Final installation¶
To install the package procars, and all its dependances, just do:
sudo pip install .
or:
sudo python setup.py install
You will then be able to use ProCARs from any directory in your computer, just as any other software.
Warning
This must be done only if you downloaded a stable version of the package, and won’t do any more changes on the scripts and modules. Indeed, by installing the package, the changes done after won’t be taken into account while running the scripts. If you plan to work on the scripts, choose the deployment installation (see above).
Note
You can always uninstall ProCARs if you want to install a new version, or work on the scripts.
Uninstalling ProCARs¶
Whatever the way you installed ProCARs, you can uninstall it by running:
sudo pip uninstall procars
Running ProCARs¶
When you have installed ProCARs (in final or development mode, see here-above for more information) you can run it from anywhere in your computer, by calling the following command:
procars_main -t tree_file -b block_file -r result_directory
where tree_file and block_file are the paths to your input files, for the phylogeny and the orthology blocks respectively. result_directory is the directory in which you want to save all ProCARs results.
If you want to work on the ProCARs code, and do not want to install the package, you must run ProCARs from its root directory (same directory as procars, bin, setup.py etc.), and use the following command:
PYTHONPATH+=. python bin/procars_main -t tree_file -b block_file -r result_directory
ProCARs file formats¶
The input files are :
- a phylogeny tree, with Newick format, and with a @ at the required ancestor node
- a block file, containing all blocks position and orientation for all extant genomes of the tree
You also have to specify a directory where you want to store all results from ProCARs run. If the specified directory does not already exist, it will be created.
Note
If you run ProCARs twice on the same result directory, it will erase the last results and replace them by the new ones.
The output is a Q-tree, corresponding to the ancestor’s reconstructed genome, composed of CARs.
Input files¶
You can find examples of those input files into the Examples directory of ProCARs.
Phylogenic tree
The phylogenic tree is at the Newick tree format. You only need to give the phylogeny, with all extant species names, but no branch lengths are needed. You also have to tag an ancestor node with a @ to specify which ancestor’s genome you want ProCARs to reconstruct. The ancestor must be a node splitting the tree into three subtrees: two ingroups and one outgroup.
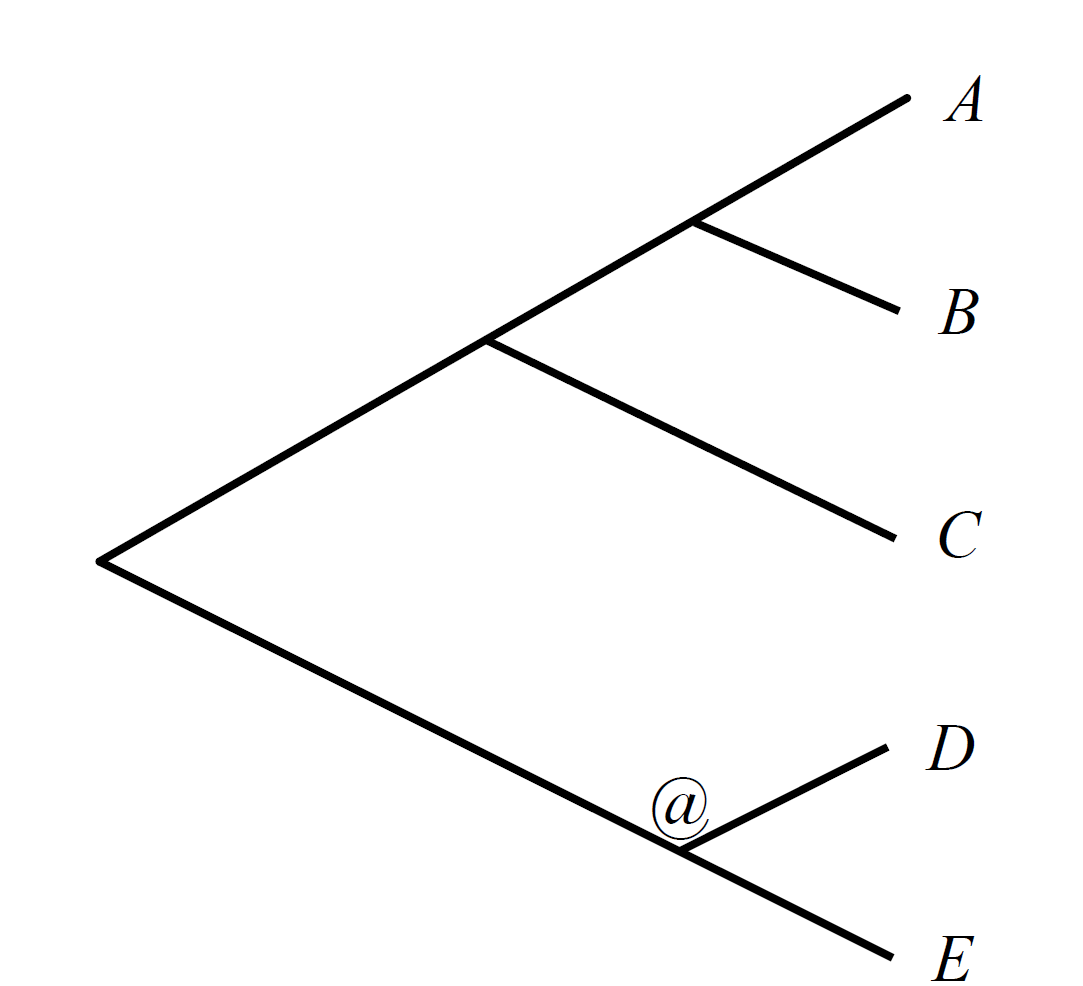
Here is an example of a species tree input:
((C,(A,B)),(D,E)@);
corresponding to this tree:
Note
If your extant genomes have long names with several words, separate the words with _ instead of a space.
Orthology blocks
The orthology blocks have the following format:
>*block_number*
*species1_name*.*chromosome_name*:*start*-*end* *orientation*
*species2_name*.*chromosome_name*:*start*-*end* *orientation*
*species3_name*.*chromosome_name*:*start*-*end* *orientation*
*species4_name*.*chromosome_name*:*start*-*end* *orientation*
>*block_number*
*species1_name*.*chromosome_name*:*start*-*end* *orientation*
*species2_name*.*chromosome_name*:*start*-*end* *orientation*
*species3_name*.*chromosome_name*:*start*-*end* *orientation*
*species4_name*.*chromosome_name*:*start*-*end* *orientation*
where start and end are the nucleotide positions of the current block on the chromosome containing it, and orientation is either + or - according to the orientation of the block on the species chromosome. For example, the following could be the beginning of an orthology block input file for the tree in the previous example:
>1
A.1:1-99 +
B.1:1-99 +
C.1:1-99 +
D.1:1-99 +
E.1:1-99 +
>2
A.1:101-199 +
B.1:101-199 +
C.1:101-199 +
D.1:101-199 -
E.1:101-199 +
Output files¶
The output is a Q-tree, corresponding to the ancestor’s reconstructed genome, composed of CARs. Each CAR is an ordered list of signed blocks. The format of a Q-tree is the following:
#CAR1
_Q block1 block2 block3 block4 ... Q_
#CAR2
_Q block1 block2 block3 block4 ... Q_
#CAR3
_Q block1 block2 block3 block4 ... Q_
where each block is a signed integer, indicating its orientation. For example:
#CAR1
_Q 1 2 3 -5 Q_
#CAR2
_Q -7 -6 4 Q_
#CAR3
_Q 8 Q_
In your result directory, you will also find a subfolder called procars_steps. In this directory, you can find all intermediate ancestor trees (S_PQtree_ancestor_ + step number of ProCARs in which the Q-tree was computed), with the same format as the output file. You will also find files describing at which step each adjacency was added, and its type (S_Adjacencies_ancestor... + step number of ProCARs at which the adjacency file was created). The adjacency files have the following format:
block1_adj1 block2_adj1 car1_adj1 car2_adj1 type_adj1 step_adj1 labels_adj1
block1_adj2 block2_adj2 car1_adj2 car2_adj2 type_adj2 step_adj2 labels_adj2
block1_adj3 block2_adj3 car1_adj3 car2_adj3 type_adj3 step_adj3 labels_adj3
where block1 block2 is the block adjacency (the two blocks involved in the adjacency added to the Q-tree) and car1 car2 is the car adjacency corresponding to the block adjacency. type_adj is the type of the given adjacency. It is either 1 for a fully conserved adjacency, 0 for a partly conserved adjacency, or 2 for a DCJ-reliable adjacency. Finally, labels_adj are indicating if the given adjacency is present (1) or absent (0) in each of the extant genomes. Hence, there are as many labels as extant genomes. Here is an example of an adjacency file, for the input tree example (5 extant genomes):
6 7 6 7 0 1 1 1 1 1 1
1 2 1 2 1 1 1 1 1 0 1
2 3 2 3 1 1 1 1 1 0 1
3 -5 3 -5 1 1 0 0 0 1 1
-4 6 -4 6 1 1 0 0 0 1 1
You will finally find other adjacency files, with a suffix _discarded.txt, or _discarded_used.txt. The first one corresponds to the conflicting adjacencies saved by a step a) of ProCARs. If, in the same step, the set of non-conflicting adjacencies is empty, ProCARs will go to the step b) (resolve conflicts) and hence use this file. At the end of this step b), the file suffixed by _discarded.txt is empty (conflicts are resolved), but its content is saved to the file suffixed by _discarded_used.txt, if you need to check which adjacencies were involved in a resolved conflict. All these adjacency files have the same format, described here-above.
Running tests¶
If you want to work on ProCARs scripts, you can use the tests provided with the software, used to check each of its functionalities. To run the tests, run, from the root of the project:
PYTHONPATH+=. py.test test/
Or, if you installed the package (final or development mode):
py.test test/