This page contains experimental datasets, results and associated scripts related to the draft paper Spaced seed design on profile HMMs for precise HTS read-mapping. This content is related to the Section 4 Experiments on seed design of the draft paper. It is divided in two parts, the section 1. Initial data preparation , and the experimental section 2. Seed design.
alignment.fasta.gz → (hmmer 3.0) → hmmmodel.hmm → (hmmread.pl) → autogen_from_hmm.txt
The initial model used is an alignment [fasta.gz] of 5435 Bacterial 16S rRNA sequences. This alignments is directly derived from a GreenGenes.lbl.gov alignment [fasta.gz] of 5441 Gold reference Bacterial 16S rRNA sequences, where 6 unaligned sequences have been cleaned up.
This alignment has first been converted into a profile hmm [hmm] with help of hmmer [tar.gz] using this command line : hmmbuild --rna --informat afa hmmmodel.hmm alignment.fasta
This profile hmm model has then been converted into a simplified probabilistic match/mismatch/(set of indels) automaton readable by iedera [dir] with help of this perl script [pl] (input and output file inlined in its code).
The probabilistic automaton obtained in section 1. Initial data preparation provides a typical model for alignments that can be drawn by aligning any two 16S bacterial rRNA sequences. As thus, it can be used to design seeds to match the full 16S rRNA alignment. We are interested (in the draft paper Spaced seed design on profile HMMs for precise HTS read-mapping) not by the full alignment window, but by all the sub-windows of this alignment to give a precise measure of the sensitivity and the coverage of the 16S rRNA when this one is being mapped with HTS reads.
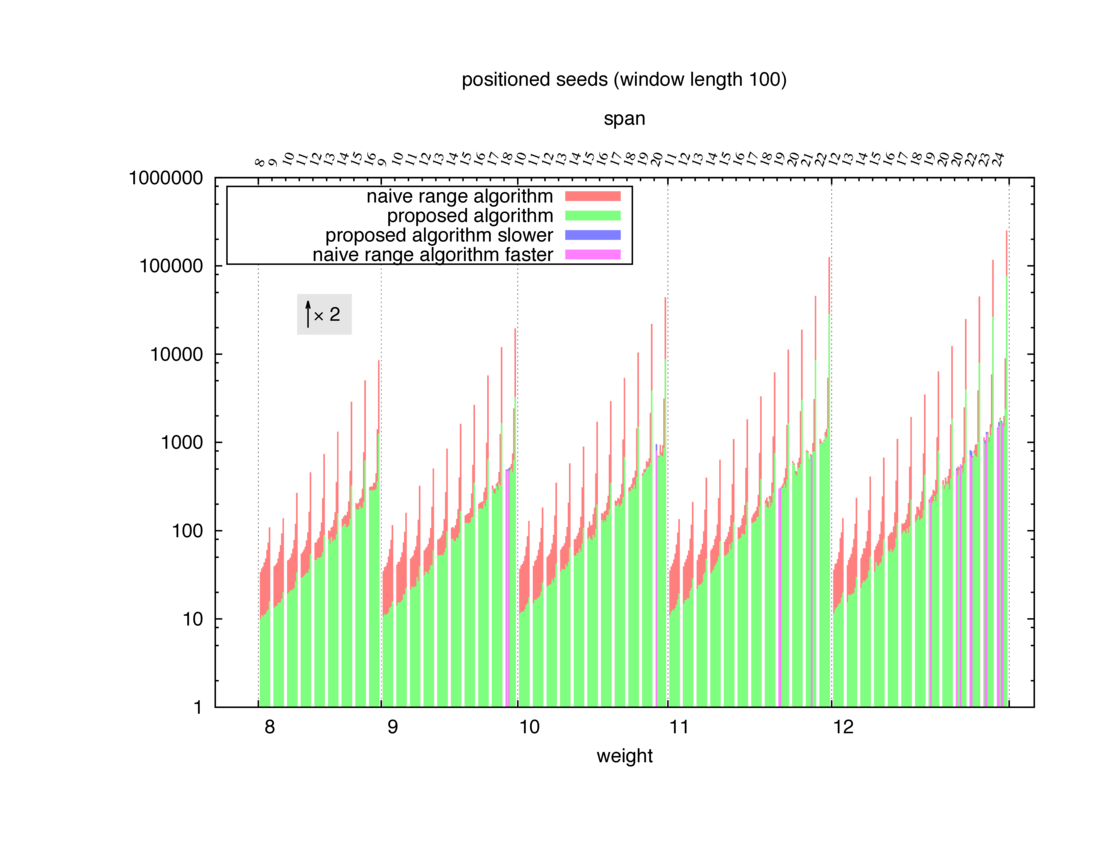
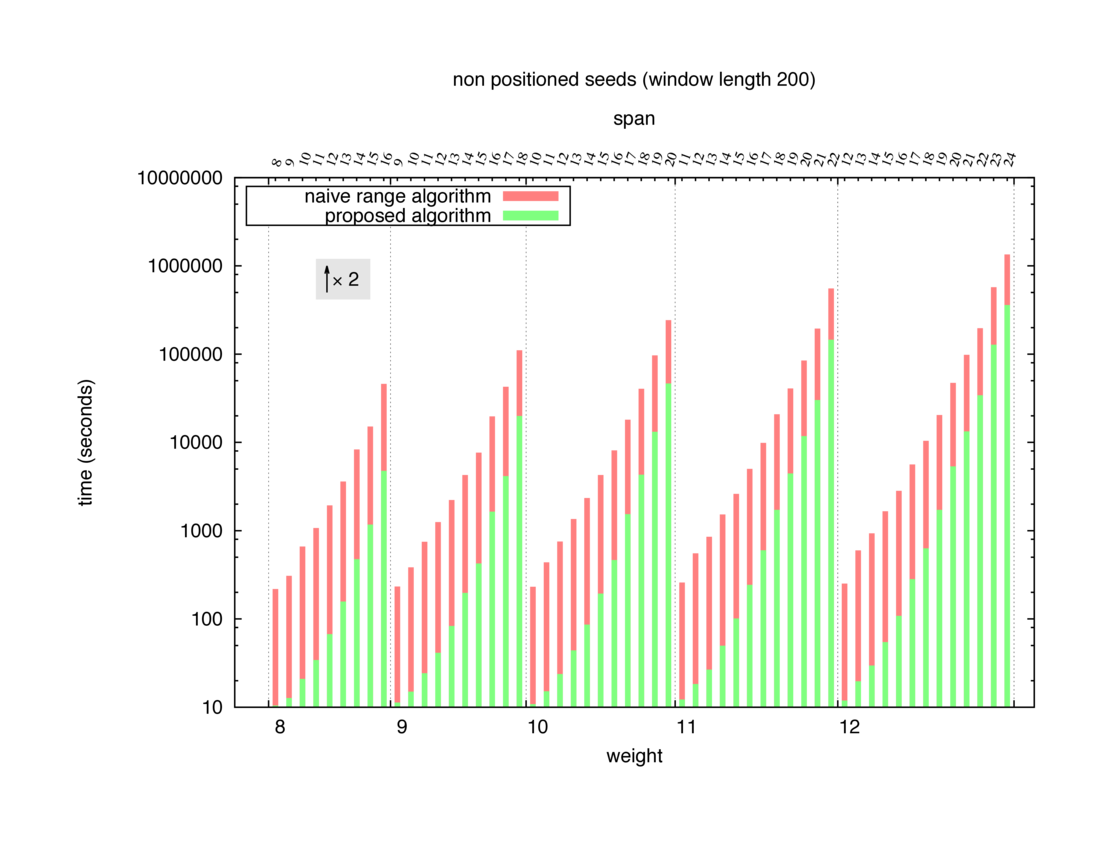
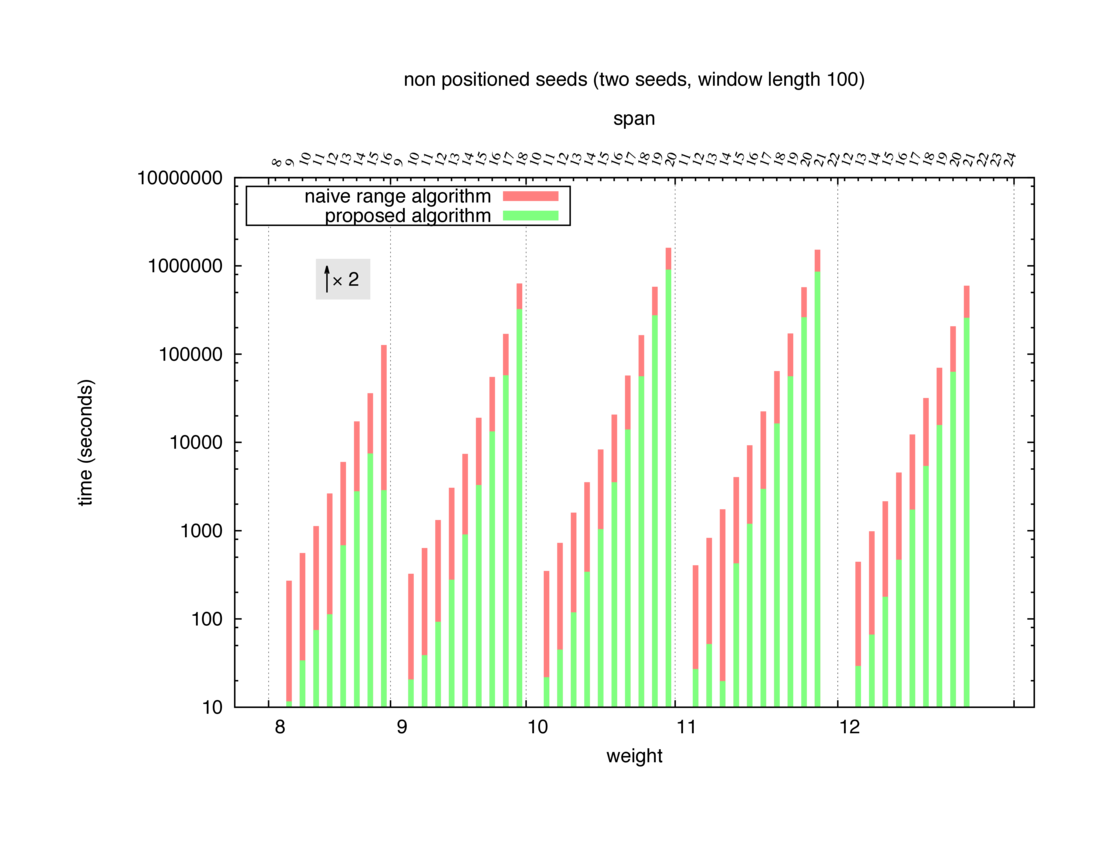
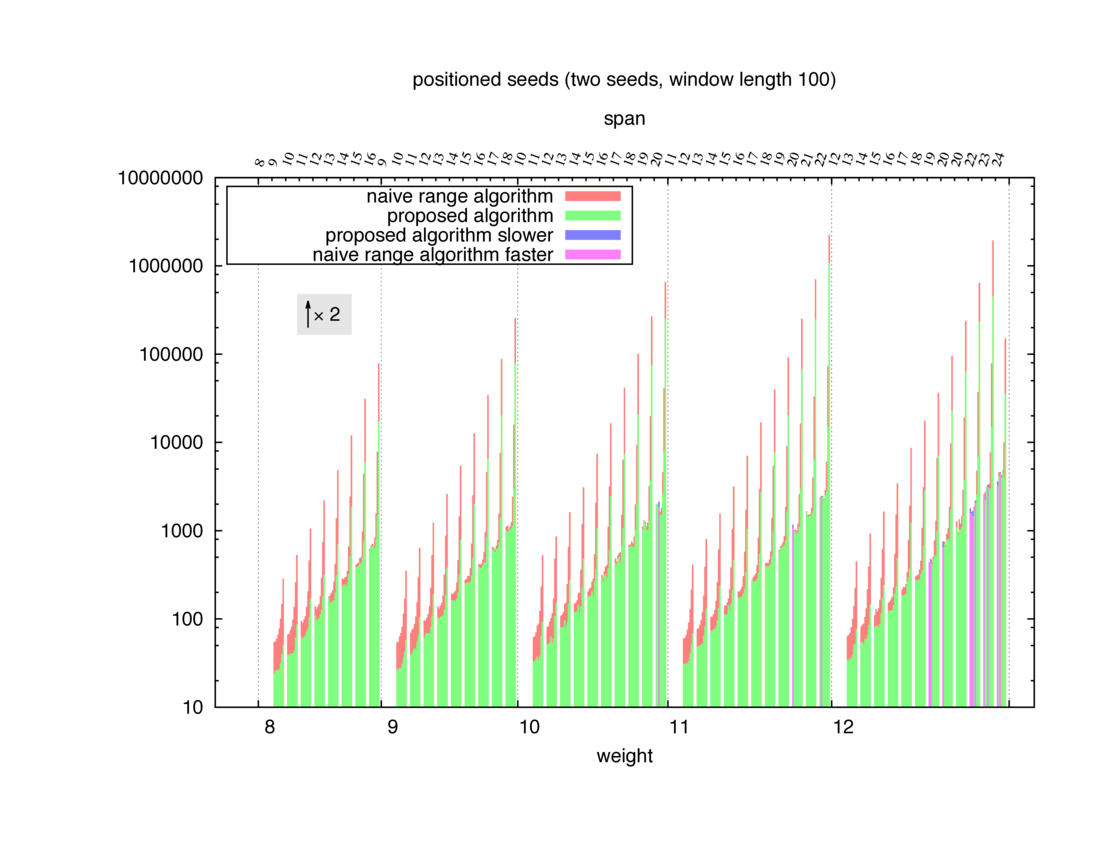
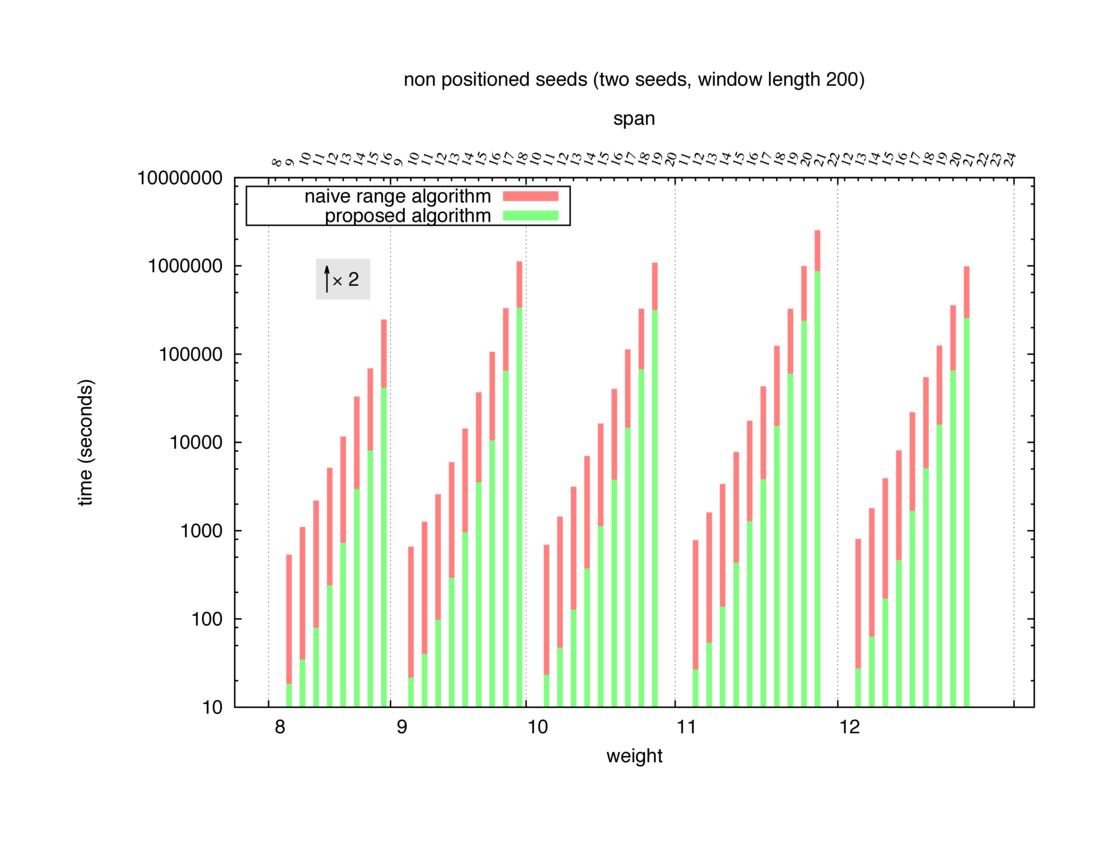
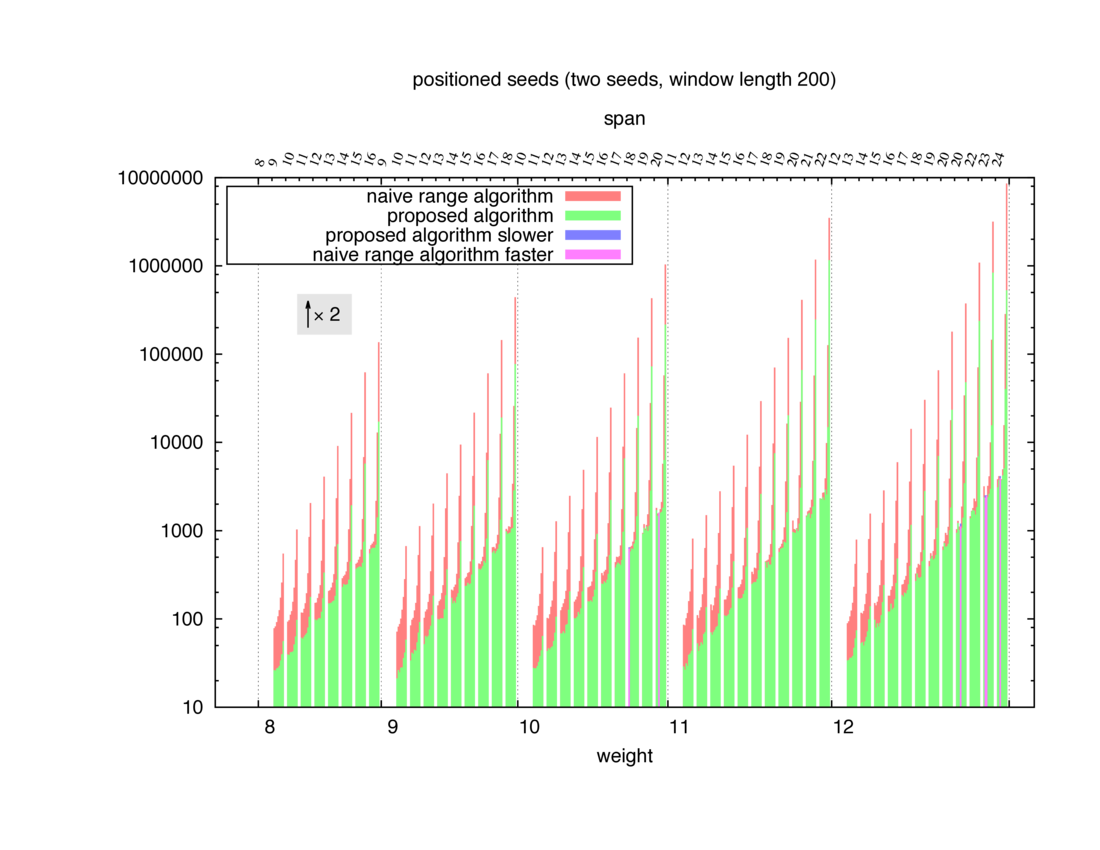
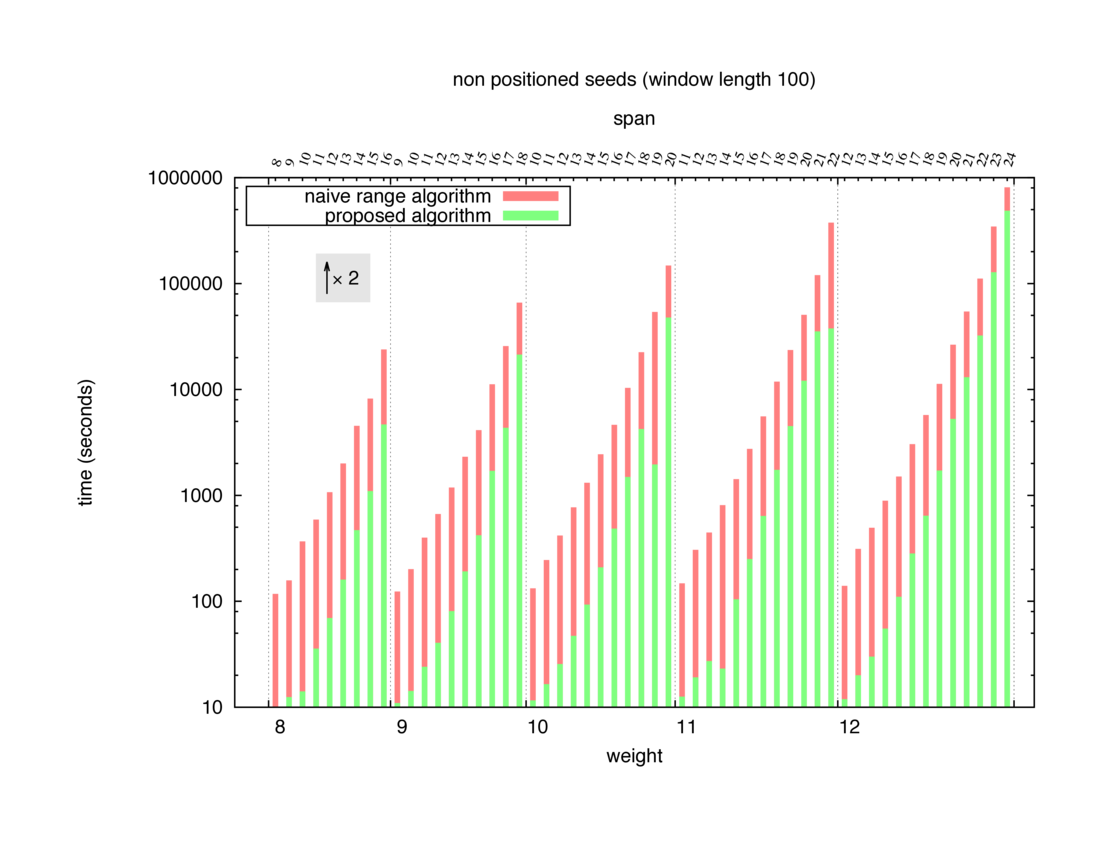
Two algorithms to compute seed sensitivity are studied in this example : one is a naive algorithm where all the sub-windows are enumerated and computed using a (vector × matrix) × matrix ... product, and the other proposed algorithm use a factorized form of a matrix × matrix ... product.
Both algorithms have been used to design 100 seeds each time of different weight and span, and the time has been measured for each set. Window size (read length) was set to 100 or 200 (two experimental sets), and seeds have been designed to be applied, either at any position of the read, either at a restricted set of positions (two experimental sets, the second with several set of positions : 10,20,40,80,160,320,640,1280). Finally, both Single seeds and Multiple seeds have been designed (two experimental sets).
To do so, a script [sh] has first been run to measure the time spent by iedera with each of the two algorithms (either the naive algorithm with the iedera-noslicer binary, otherwise the proposed algorithm with the iedera-slicer binary) : this script generates several csv files, one per experiment [dir]. The two(/three) first csv columns are organized to give the weight w, the span s, (and possibly the number of positions q), for the set of seeds tested. The two last csv columns give the time spent by iedera-noslicer and iedera-slicer when computing this set of reads. At the end, these csv files have then been plotted by several gnuplot scripts [dir] to provide a visual comparison of the time spent by iedera-noslicer and iedera-slicer [dir].