This page is a user manual for miRkwood ab initio web application.
- Input form
- Enter query sequence
- Parameters
- Submit the job
- Result page
- Export
- Tabular format (CSV)
- FASTA
- dot-bracket format
(plain sequence + secondary structure) - Full report in ORG format
- GFF format
- HTML report
- Header
- Thermodynamic stability
- Conservation of the mature miRNA
Input form
The input form of miRkwood has three main sections:
- Enter query sequence,
- Parameters: select additional annotation criteria for the miRNA precursors,
- Submit the job.
Results are displayed on a new page.
Enter query sequence
miRkwood input is (Multi) FASTA format. Lower-case and upper-case letters are both accepted, as well as T/U. Other characters, such as N, R, Y,… are prohibited.
You can either paste or upload a file. The maximum size for a submission is 100 000 nt.
Scan both strands: miRkwood normally analyses data in forward direction only. Checking this option will cause the program to search both the forward and reverse complement strands.
Mask coding regions: This option allows selecting non-coding sequences in the input data by masking putative coding regions. It consists in a BlastX search against the protein sequences from the chosen species (E-value=1E-5). Currently available species are: Arabidopsis thaliana (TAIR, V10), Medicago truncatula (Medicago truncatula genome project, Mt4.0) and Oryza sativa (Rice genome annotation project, V7.0).
Filter tRNA/rRNAs: If this option is checked, tRNAs and rRNAs are filtered out from the input data. The prediction of tRNA genes is done with tRNAscan-SE. The prediction od rRNA genes is done with RNAmmer, with option -S euk -m tsu, lsu, ssu.
Run with an example: We provide the user with a sample sequence. This is a 268 nt expressed sequence from Salvia sclarea (Legrand et al., 2010) that lacks any annotation and is not present in miRBase.
Parameters
miRkwood folds the input sequence to identify miRNA precursor secondary structures with RNALfold. The maximal size is 400nt and the minmal size is 70nt. This gives a set of candidate pre-miRNAs. For each candidate pre-miRNA, it is possible to calculate additional criteria that help to bring further evidence to the quality of the prediction and to distinguish accurate miRNA precursors from pseudo-hairpins.
When all options are unchecked, no criteria is applied, and the list of predictions is exactly the list of all candidate pre-miRNAs obtained with the folding step.
Select only sequences with MFEI < -0.6: MFEI is the minimal folding free energy index. It is calculated by the following equation:
MFEI = [MFE / sequence length x 100] / (G+C%)
where MFE (minimal free energy) denotes the negative folding free energies of a secondary structure, and is calculated using the Matthews-Turner nearest neighbor model implemented in RNAeval. When checked, this option removes all candidate pre-miRNAs with an MFEI greater than or equal to -0.6. Indeed, more than 96% of plant miRBase precursors have an MFEI smaller than -0.6, whereas pseudo-hairpins show significantly larger values of MFEI. Default: checked.
Compute thermodynamic stability: The significance of the stability of the sequence can also be measured by comparison with other equivalent sequences. Bonnet et al have established that the majority of the pre-miRNA sequences exhibit a MFE that is lower than that for shuffled sequences. We compute the probability that, for a given sequence, the MFE of the secondary structure is different from a distribution of MFE computed with 300 random sequences with the same length and the same dinucleotide frequency. Default: unchecked.
Flag conserved mature miRNAs: Some families of mature miRNAs are highly conserved through evolution. In this case, it is possible to localize the mature miRNA within the pre-miRNA by similarity. For that, we compare each sequence with the mature miRNAs of plant (Viridiplantae) deposited in miRBase (Release 21). We select alignments with at most three errors (mismatch, deletion or insertion) against the full-length mature miRNA and that occur in one of the two arms of the stemloop. Moreover, this alignment allows to infer a putative location for the miRNA within the precursor. The putative location obtained is then validated with miRdup, that assesses the stability of the miRNA-miRNA* duplex. Here, it was trained on miRBase Viridiplantae v21. Default: checked.
Submission
Each job is automatically assigned an ID.
Job title: It is possible to identify the tool result by giving it a name.
Email address: You can enter your email address to be notified when the job is finished. The email contains a link to access the results for 2 weeks.
Result page
Results are summarized in a two-way table. Each row corresponds to a pre-miRNA, and each column to a feature. By default, results are sorted by sequence and then by position. It is possible to have them sorted by quality (see definition). You can view all information related to a given prediction by clicking on the row (see section HTML Report).
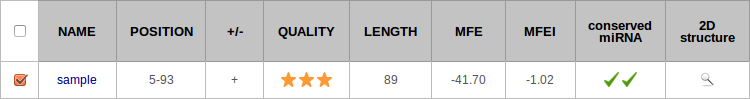
Name: Name of the original sequence, as specified in the heading of the FASTA format.
Position: Start and end positions of the putative pre-miRNA in the original sequence.
+/- (option): Strand, forward or reverse complement.
Quality: The quality is a distinctive feature of miRkwood. It is a combination of all other criteria described afterwards, and allows to rank the predictions according to the significance, from zero- to three- stars. It is calculated as follows.
- MFEI < -0.8: add one star. This MFEI threshold covers 83% of miRBase pre-miRNAs, whereas it is observed in less than 13% of pseudo hairpins.
- Existence of a conserved miRNA in miRBase (alignment): add one star. We allow up to three errors in the alignment with mature miRBase, which corresponds to an estimated P-value of 3E-2 for each pre-miRNA. Alignments with 2 errors or less have an estimated P-value of 4E-3.
- The location of the mature miRNA obtained by alignment is validated by miRdup: add one star.
MFE: Value of the minimal free energy (computed with RNAeval).
MFEI: Value of the MFEI (see definition).
Shuffles (option): Proportion of shuffled sequences whose MFE is lower than the MFE of the candidate miRNA precursor (see Compute thermodynamic stability). This value ranges between 0 and 1. The smaller it is, the more significant is the MFE. We report pre-miRNA stemloops for which the value is smaller than 0.01, which covers more than 89% of miRBase sequences. Otherwise, if the P-value is greater than 0.01, we say that it is non significant, and do not report any value.
miRBase alignment (option): This cell is checked when an alignment between the candidate sequence and miRBase is found (see Flag conserved mature miRNAs). It is doubled checked when the location of the candidate mature miRNA is validated by miRdup. The alignments are visible in the HTML report.
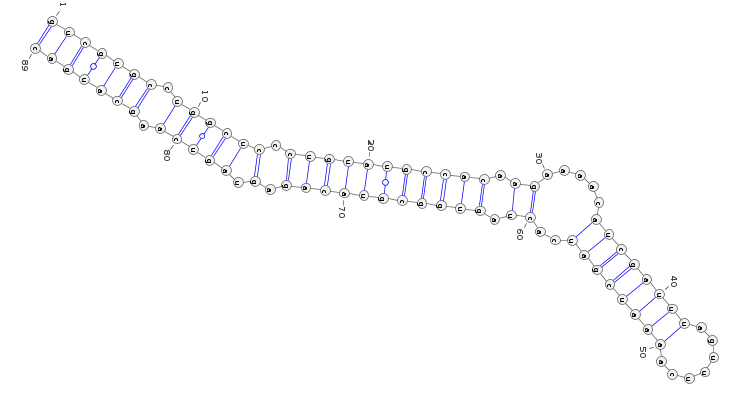
2D structure : You can drag the mouse over the zoom icon to visualize the stemloop structure of the pre-miRNA. The image is generated with Varna.
Export
Results, or a selection of them, can also be exported to a variety of formats, and saved to a local folder for further analyses.
Export is limited to 200 candidates at a time. You may either export all candidates ("Select all" button), or perform several successive exports.
Tabular format (CSV): It contains the same information as the result table, plus the FASTA sequences and the dot-bracket secondary structures. The CSV format is supported by spreadsheets like Excel. See more information on CSV.
FASTA format: This is the compilation of all pre-miRNA sequences found in FASTA format. The header of the FASTA format contains the initial name of the sequence, as well as the positions and the strand of the predicted pre-miRNA.
Dot-bracket format: This is the compilation of all pre-miRNA sequences found, together with the predicted secondary structure. The first line contains a FASTA-like header. The second line contains the nucleic acid sequence. The last line contains the secondary structure, that is given as a set of matching brackets. A base pair between bases i and j is represented by a "(" at position i and a ")" at position j. Unpaired bases are represented by dots (see more explanation on Vienna website).
>sample__5-93, stemloop structure gucgugccuggcucccuguaugccacaagaaaacaucgauuuaguuucaaaaucgaucacuaguggcguacagaguagucaagcaugac (((((((.((((((.(((((((((((.((.....((((((((.......))))))))..)).))))))))))).).))))).)))))))
Full report in ORG format: This is an equivalent of the HTML report, and contains the full report of the predictions.
GFF format : General annotation format, that displays the list of positions of pre-miRNA found (see more explanation on Ensembl documentation)
##gff-version 3 # miRNA precursor sequences found by miRkwood have type 'miRNA_primary_transcript'. # Note, these sequences do not represent the full primary transcript, # rather a predicted stemloop portion that includes the precursor. sample miRkwood miRNA_primary_transcript 5 93 . + . Name=preMir_sample__5-93
Following the convention of miRBase, we consider that hairpin precursors have type miRNA_primary_transcript even if they are shorter than the primary transcript.
HTML Report
The HTML report contains all information related to a given predicted pre-miRNA.
Header
The report begins with the following information.
Name: Name of the initial sequence, as specified in the heading of the FASTA format.
Position: Start and end positions of the putative pre-miRNA in the original sequence. The length is indicated in parentheses.
Strand: + (forward) or - (reverse complement).
GC content: Percentage of bases that are either guanine or cytosine.
Sequence (FASTA format): Link to download the sequence.
Stemloop structure : Link to download the secondary structure in dot-bracket format (see definition).
Optimal MFE secondary structure: If the stemloop structure is not the MFE structure, we also provide a link to download the MFE structure.
Alternative candidates (dot-bracket format): This is the set of stemloop sequences that overlap the current prediction. The choice between several alternative overlapping candidate pre-miRNAs is made according to the best MFEI.
The stemloop structure of the miRNA precursor is also displayed with Varna.
Thermodynamics stability
MFE: Value of the Minimum Free Energy (computed by RNAeval).
AMFE: Value of the adjusted MFE: MFE ÷ (sequence length) × 100
MFEI: Value of the minimum folding energy index (see definition).
Shuffles: Proportion of shuffled sequences whose MFE is lower than the MFE of the candidate miRNA precursor (see Compute thermodynamic stability). This value ranges between 0 and 1. The smaller it is, the more significant is the MFE. We report pre-miRNA stemloops for which the value is smaller than 0.1, which covers more than 89% of miRBase sequences. Otherwise, if the P-value is greater than 0.1, we say that it is non significant, and do not report any value.
Conservation of the miRNA
All alignments with miRBase are reported and gathered according to their positions.
query 5 ugccuggcucccuguaugcca 25
|||||||||| |||||||||
miRBase 1 cgccuggcuccuuguaugcca 21
miRBase sequence: ppt-miR160h
query is the user sequence, and miRBase designates the mature miRNA (or the miRNA*) found in miRBase. It is possible to access the corresponding miRBAse entry by clicking on the link under the alignment. The report also indicates whether the location is validated by miRdup. Finally, we provide an ASCII representation of the putative miRNA within the stemloop precursor.
c - c a aaaac ag
gucgugc uggcu c cuguaugccac ag aucgauuu u
||||||| ||||| | ||||||||||| || |||||||| u
caguacg acuga g gacaugcggug uc uagcuaaa u
a u a a ac--- ac