Translation dependent score matrices
The translation-dependent scoring scheme we designed incorporates information about possible mutational patterns for coding sequences, based on a codon substitution model, with the aim of filtering out alignments between sequences that are unlikely to have common origins.
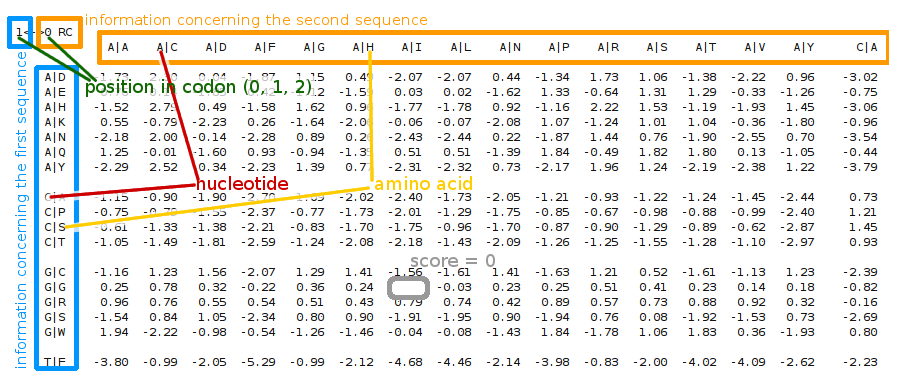
With the aim of retracing the sequence's evolution and revealing which base substitutions are more likely to occur within a given codon, our scoring system targets pairs of triplets (n, p, a), were n is a nucleotide, p is its position in the codon, and a is the amino acid encoded by that codon, thus differentiating various contexts of a substitution.
For detailed information about the actual computation method of the translation dependent scores, please refer to [1] and [2].
Sample matrices for several evolutionary distances
The evolutionary distances reflect the expected number of mutations per codon: 00.01, 00.10, 00.30, 00.50, 00.70, 01.00, empirical (score matrix based on an empirical codon substitution model).
How to read the score matrix files
[1] Gîrdea, M. and Noé, L. and Kucherov, G.: Back-translation for discovering distant protein homologies, in Proceedings of WABI 2009, Philadelphia, September 12 – 13, 2009
[2] Gîrdea, M. and Noé, L. and Kucherov, G.: Back-translation for discovering distant protein homologies in the presence of frameshift mutations, Algorithms for Molecular Biology, Volume 5, January 2010