How does TnSeek work ?
TnSeek is designed to efficiently handleTn-seq data coming from multiple strains and growth conditions.
It takes as inputs:
- a set of raw reads (specifically illumina reads),
- the list of reference genomes along with their annotation,
- an orthology table between genes from the strains.
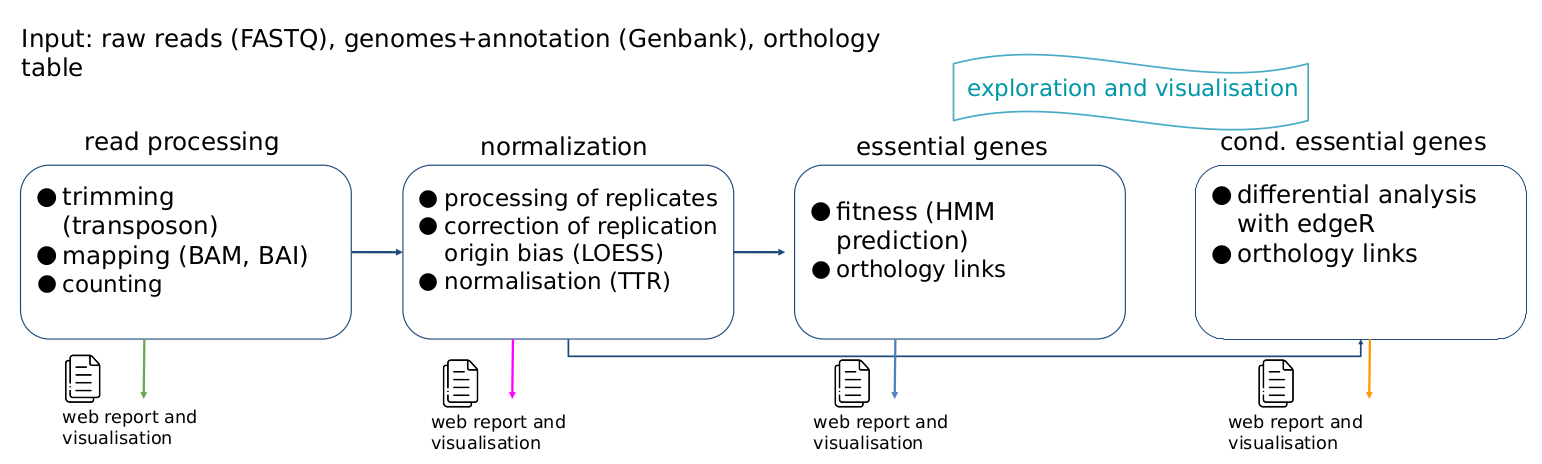
The method involves four steps.
Step 1 : read processing (trimming, mapping and counting)
Reads are first filtered out in order to select only those containing the transposon sequence. This sequence is trimmed from reads with cutadapt, and reads with required length were mapped on the reference genome with bowtie (end-to-end alignment, no mismatch). Only reads with one single alignment and whose 3' end aligns with a "TA" dinucleotide are selected. Other reads are discarded. This allows us to construct the raw count tables.
Step 2 : normalization (processing of replicates, correction of replication origin bias, normalisation)
We first apply a LOESS normalization (local weighted regression) in order to correct the replication origin bias. Non-biological variability between tables was then eliminated by TTR normalization (Trimmed Total Reads). At the end of this step, we get one normalized count table per strain and per condition, together with several quality control metrics.
Step 3 : essential genes (fitness).
For each strain, we identify essential genes from the count tables using TRANSIT's HMM method.
Step 4 : conditionally essential genes (differential analysis)
Experimental conditions are compared to control conditions using TRANSIT's RESAMPLING method.
TnSeek is fully-automated and ensures complete reproducibility of the results. Moreover, TnSeek provides a wide range of customizable parameters that can be tailored to the user's specific requirements : transposon sequence, minimal read length, normalization, etc.
Tnseek comes with a user-friendly interface, and facilitates result interpretation by generating a comprehensive report that can be conveniently accessed through any web browser. This report enables users to navigate through the analysis process step by step, accompanied by a diverse range of figures and graphs. For a holistic understanding, all results are summarized in global tables at the strain level, providing a comprehensive overview. Additionally, Tnseek offers additional insights into essential genes and conditionally essential genes across species with orthology links between strains, thus facilitating comparative analysis. Lastly, Tnseek includes a visualization tool dedicated to investigating individual genes.
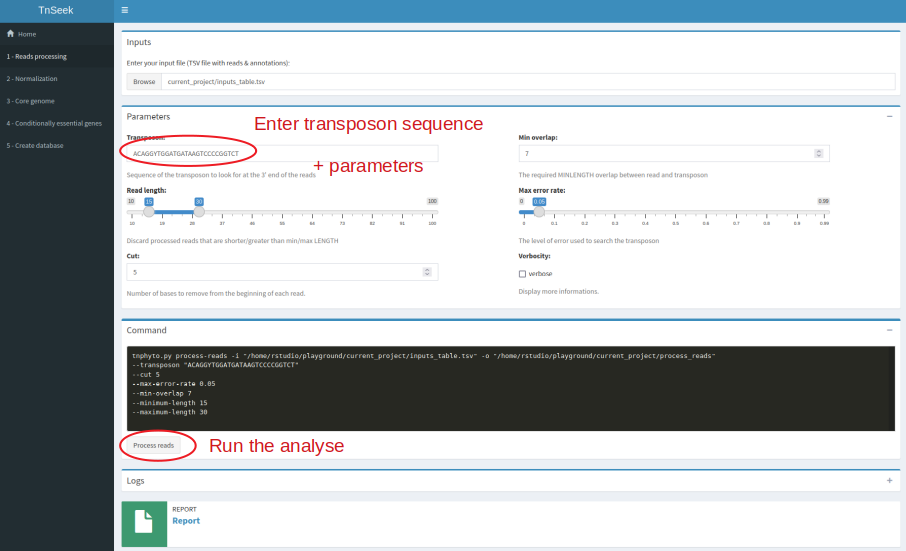
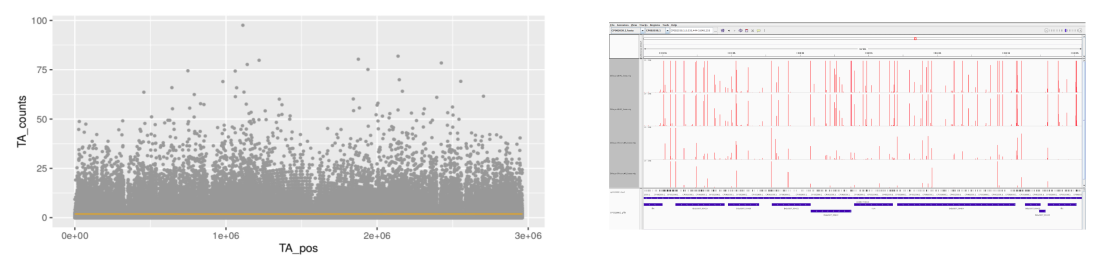
The core of TnSeek is implemented in Python and R. The graphical user interface, that is to say, the web application for exploration and visualization are in R Shiny and SQLite. The report is in R markdown.