This page is a user manual for the miRkwood small RNA-seq web application.
If this is your first time using miRkwood, we would suggest visiting our quick start guide before.
Input form
Upload your set of reads
The input is a set of reads produced by deep sequencing of small RNAs and then mapped to a reference genome. For that, you should organize your data in a BED file. See the detailed instructions on how to build this file, or grab a sample file below.
You will find more information on this sample file in our quick start guide.
Select an assembly
miRkwood currently proposes 12 assemblies, which are listed below.
- Arabidopsis lyrata - v1.0 - GFF: Phytozome
- Arabidopsis thaliana - TAIR 10 - GFF: Phytozome
- Brassica napus - Bnapus 4.1 - GFF: brassicadb
- Brassica rapa - Brapa 1.1 - GFF: brassicadb
- Glycine max - v1.0.23 - GFF: Ensembl
- Lotus japonicus - Lj2.5 - GFF: kazusa.or.jp
- Medicago truncatula - JCVI_Mt3.5.2 - GFF: JCVI
- Oryza sativa - MSU7 - GFF: Phytozome
- Populus trichocarpa - JGI_Poptr2.0 - GFF: Phytozome
- Solanum lycopersicum - SL2.4 - GFF: Phytozome
- Sorghum bicolor - Sorbi1 - GFF: PlantGDB
- Vitis vinifera - Genoscope-20100122 - GFF: Phytozome
When available, the assembly is supplemented by two GFF files. The first one contains the genome coordinates of annotated CDSs, tRNAs, rRNAs and snoRNAs, and is used to apply masking options described in section Parameters. The source of this file is indicated above, case by case. The other GFF file compiles all miRNAs and precursors of miRNAs available in MiRBase 21 and is used to detect known miRNAs that are expressed in the sequencing data.
Parameters
miRkwood comes with a series of options, that allow to customise the search and enhance the results. These options can be divided in two main types: parameters concerning the processing of the sequence reads and parameters concerning the secondary structure of the miRNA precursor. Note that they only apply to novel miRNAs. For known miRNAs, the user can rely on the additional information delivered by miRBase and make up his/her own mind.
Parameters for the processing of the read data
The first step of miRkwood is to locate signals into the set of mapped reads. This is performed by scanning the set of reads and detecting statistically significant clusters of reads. To this end, it is advised to filter out the data beforehand with the following options.
Mask coding regions: This option allows selecting reads that are aligned to non-coding sequences. The selection is performed with the GFF annotation file. All reads that intersect a CDS feature are removed. Default: checked.
Filter out tRNA/rRNA/snoRNA: When this option is checked, products from tRNA, rRNA and snoRNA degradation are filtered out from the input reads. This task is performed based on the existing annotation provided in the GFF annotation file. All reads that intersect a tRNA, rRNA, or snoRNA feature are removed. Default: checked.
Remove multiply mapped reads: All reads that are mapped to more than 5 loci on the reference sequence are discarded. This allows to avoid spurious predictions due to transposons. Default: checked.
Parameters for the secondary structure of the hairpin precursor
After cluster detection, miRkwood aims at determining which sequences can fold into a stemloop structure. This gives a set of candidate precursors of miRNAs. For each candidate, it is possible to calculate additional criteria that help to bring further evidence to the quality of the prediction and to distinguish accurate miRNA precursors from pseudo-hairpins.
Select only sequences with MFEI < -0.6: MFEI is the minimum folding free energy index, and expresses the thermodynamic stability of the precursor. It is calculated by the following equation:
MFEI = [MFE / sequence length x 100] / (G+C%)
where MFE (minimum free energy) denotes the negative folding free energies of a secondary structure, and is calculated using the Matthews-Turner nearest neighbor model implemented in RNAeval. When checked, this option removes all candidate pre-miRNAs with an MFEI greater than or equal to -0.6. Indeed, more than 96% of miRBase precursors have an MFEI smaller than -0.6, whereas pseudo-hairpins show significantly larger values of MFEI. Default: checked.
Compute thermodynamic stability: The significance of the stability of the sequence can also be measured by comparison with other equivalent sequences. Bonnet et al have established that the majority of the pre-miRNA sequences exhibit a MFE that is lower than that for shuffled sequences. We compute the probability that, for a given sequence, the MFE of the secondary structure is different from a distribution of MFE computed with 300 random sequences with the same length and the same dinucleotide frequency. Default: unchecked.
Flag conserved mature miRNAs: This option permits to check if the predicted miRNA belongs to some known miRNA family. For that, we compare the sequence of the precursor with the database of mature miRNAs of plant (Viridiplantae) deposited in miRBase (Release 21). We select alignments with at most three errors (mismatch, deletion or insertion) against the full-length mature miRNA and that occur in one of the two arms of the stemloop. Moreover, this alignment allows to infer a putative location for the miRNA within the precursor. This location is then validated with miRdup, that assesses the stability of the miRNA:miRNA* duplex. Here, it was trained on miRbase Viridiplantae V21. Default: checked.
Filter out low quality hairpins: When this option is checked, hairpins found by miRkwood with a global score of 0 and no alignment with miRbase are discarded. Default: checked.
Submission
Each job is automatically assigned an ID.
Job title. It's possible to identify the tool result by giving it a name.
Email Address. You can enter your email address to be notified when the job is finished. The email contains a link to access the results for 2 weeks.
Results page
Results overview
This page has two main parts. The first one (Options summary) is simply a summary of your job parameters. The other one (Results summary) provides the detailed results.
Total number of reads (unique reads): This is the total number of reads in your initial file. The number of unique reads (obtained after merging identical reads) is indicated in parentheses.
CoDing Sequences: This is the number of reads that have been discarded by the option Mask coding regions. You can list them by clicking on the download link.
rRNA/tRNA/snoRNA: This is the number of reads that have been discarded by the option Filter out tRNA/rRNA/snoRNA. You can list them by clicking on the download link.
Multiply mapped reads: This is the number of reads that have been discarded by the option Remove multiply mapped reads. You can list them by clicking on the download link.
Orphan cluster of reads: An orphan cluster is a short region in the genome that is enriched with aligned reads but that shows no secondary structure compatible with a hairpin. You can obtain the list of the such clusters by clicking on the download link (BED file).
Orphan hairpins: An orphan hairpin is a candidate with a global score of 0 and showing no conservation with miRBase. By default, if you select the option "filter out low quality hairpins", such hairpins will be discarded automatically and you can obtain the list by clicking on the download link (BED file).
Unclassified reads: Unclassified reads are isolated reads, that do not belong to any cluster or any orphan hairpin, or do not fall in any annotated region.
Known miRNAs: This is the number of loci annotated as microRNA precursors in miRBase that intersect with reads from the BED file. You can display detailed results by clicking on the link see results. See section Known miRNAs.
Novel miRNAs: This is the number of miRNAs found by miRkwood that have not been previously reported in miRbase. You can display detailed results by clicking on the link see results. See section Novel miRNAs.
Known miRNAs
Known miRNAs are miRNAs that are already present with their precursor in the miRBase database (version 21). We consider that a known microRNA is found in the data as soon as there is at least one read on the precursor sequence. The quality score (see definition below) helps to determine which are the best candidates.
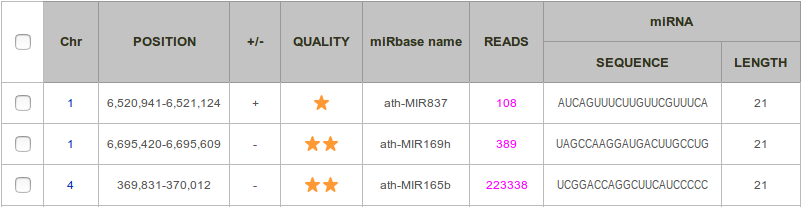
The list of all known miRNAs found is displayed in a two-way table. Each row corresponds to a pre-miRNA, and each column to a feature. By default, results are sorted by sequence and then by position. It is possible to have them sorted by quality (see definition below). You can view all information related to a given prediction by clicking on the row (see section HTML Report).
Chr: Number of the chromosome.
Position: Start and end positions of the miRNA precursor, as documented in miRBase.
+/- : Strand, forward (+) or reverse (-).
Quality: This score measures the consistency between the distribution of reads along the locus and the annotation provided in miRbase. It ranges between 0 and 2 stars, and is calculated as follows.
- the locus contains more than 10 reads: add one star.
- more than half of the reads intersect either with the miRNA or the miRNA*: add one star.
miRBAse name: miRBase identifier.
Reads: Number of reads included in the locus.
miRNA Sequence: Sequence of the miRNA.
miRNA Length: Length of the miRNA.
Novel miRNAs
Novel miRNAs are miRNAS that are not reported in miRBase. The prediction is supported by the presence of a stemloop secondary structure, a significant read coverage and read distribution.
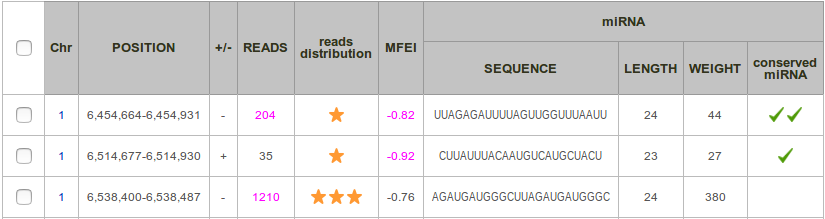
Each row corresponds to miRNA precursor, and each column to a feature. By default, results are sorted by sequence and then by position.
It is possible to have the results sorted by quality. The quality is the sum of four values:
- the existence of a miRNA sequence,
- the validation of the miRNA/miRNA* duplex by miRdup,
- the score of reads distribution,
- the value of the MFEI of the precursor secondary structure (<-0.8) (see definitions below).
You can view all information related to a given prediction by clicking on the row (see section HTML Report).
Chr: Number of the chromosome.
Position: Start and end positions of the putative miRNA precursor in the original sequence in 1 based notation (consistently to the GFF format).
+/-: Strand, forward (+) or reverse (-).
Reads: Total number of reads included in the locus.
Reads distribution: This score, ranging from 0 to 3-stars, allows to qualify the pattern of reads mapping to a putative microRNA precursor. It aims at determining if this distribution of reads presents a typical 2-peaks profile, corresponding to the guide miRNA and the miRNA* respectively.
- Number of reads: The locus has either at least 10 reads mapping to each arm, or at least 100 reads mapping in total.
- Precision of the precursor processing : At least 75% of reads start in a window [-3,+3] centered around the start position of the miRNA, or [-5,+5] on the opposite arm of the stemloop.
- Presence of the miRNA:miRNA* duplex: There is at least one read in the window [-5,+5] on the strand of the miRNA*.
Each criterion contributes equally to the overall ranking, and adds one star.
MFEI: This is the minimum folding free energy index. This value expresses the thermodynamic stability of the precursor and is calculated by the following equation:
MFEI = [MFE / sequence length x 100] / (G+C%)
where MFE is the minimum free energy of the secondary structure (computed with RNAeval) When the MFEI is < -0.8, then the value is displayed in purple, indicating a significantly stable hairpin. This MFEI threshold covers 83% of miRBase miRNA precursors, whereas it is observed in less than 13% of pseudo hairpins.
Shuffles (option): proportion of shuffled sequences whose MFE is lower than the MFE of the candidate miRNA precursor (see paragraph Compute thermodynamic stability). This value ranges between 0 and 1. The smaller it is, the more significant is the MFE. We report pre-miRNA stemloops for which the value is smaller than 0.01, which covers more than 89% of miRBase sequences. Otherwise, if the P-value is greater than 0.01, we say that it is non significant, and do not report any value.
miRNA Sequence: Sequence of the miRNA. It is the sequence of the most common read, with a frequency of at least 33%.
miRNA Length: Length of the miRNA (when existing).
miRNA Weight: Depth of the miRNA read (when existing) divided by the number of possible alignments of this read in the genome.
Conserved miRNA (option): This cell is checked  when an alignment between the miRNA precursor sequence and plant mature miRNA database of miRBase is found
(see paragraph Flag conserved mature miRNAs).
It is doubled checked
if at least 40% of reads overlap with the miRBase sequence.
The alignments are visible in the HTML report.
when an alignment between the miRNA precursor sequence and plant mature miRNA database of miRBase is found
(see paragraph Flag conserved mature miRNAs).
It is doubled checked
if at least 40% of reads overlap with the miRBase sequence.
The alignments are visible in the HTML report.
Export
Results, or a selection of them, can be exported to a variety of formats, and saved to a local folder for further analyses.
GFF: General annotation format, that displays the list of positions of pre-miRNA found (see more explanation on Ensembl documentation).
FASTA: This is the compilation of all pre-miRNA sequences found.
Dot-bracket notation: This is the compilation of all pre-miRNA sequences found, together with the predicted secondary structure. The secondary structure is given as a set of matching parentheses (see more explanation on Vienna website).
CSV (comma separated value): It contains the same information as the result table, plus the FASTA sequences and the dot-bracket secondary structures. This tabular format is supported by spreadsheets like Excel.
ORG: This is an equivalent of the HTML report, and contains the full report of the predictions. This file can be easily edited by the user.
Reads cloud: This archive is a compilation of all reads clouds. Each reads cloud is a text file that summarizes all information available for a potential precursor: positions, sequence, secondary structure, existence of an alignment with miRbase, distribution of mapped reads. It can easily be parsed.
HTML report
The HTML report contains all information related to a given predicted pre-miRNA.
HTML report for known miRNAs
- miRBase name: miRBase identifier, and link to access the miRBase entry.
- Chromosome: Name of the chromosome
- Position: Start and end positions of the pre-miRNA, such as indicated in miRBase in 1-based notation (consistently with the GFF format). The length is indicated in parentheses.
- Strand: + (forward) or - (reverse).
- G+C content: Percentage of bases that are either guanine or cytosine.
- miRNA sequence: Sequence of the predicted miRNA.
- Sequence (FASTA format): Link to download the sequence of the precursor.
- Stemloop structure: Link to download the secondary structure in dot-bracket format. The first line contains a FASTA-like header. The second line contains the nucleic acid sequence. The last line contains the set of associated pairings encoded by brackets and dots. A base pair between bases i and j is represented by a '(' at position i and a ')' at position j. Unpaired bases are represented by dots.
- Alternative candidates: This is the set of stemloop sequences that overlap the current prediction. The choice between several alternative overlapping candidate pre-miRNAs is made according to the best MFEI.
> 1:234009-234092,-, stemloop structure GAAAUGAUGCGCAAAUGCGGAUAUCAAUGUAAAUCAGGGAGAAGGCAUGAUAUACCUUUAUAUCCGCAUUUGCGCAUCAUCUCU ((.(((((((((((((((((((((.((.(((.((((.(.......).)))).))).)).))))))))))))))))))))).)).
Quality: It details the computation of the quality score as explained in section Known miRNAs.
Reads
- Number of reads: The total number of reads included in the locus.
- Reads length distribution: This plot gives the sequence length distribution for all reads in the precursor locus.
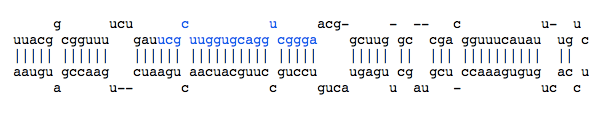
- Reads cloud: This is a visual representation of all reads included in the locus.
Each ********** string is a unique read. Its length and its depth (its number of occurrences in the set of reads) are reported at the end of the dotted line. <------miRBase------> indicates the positions of the miRNA referenced in miRBase.
Locus : 1:234009-234092
Strand : -
GAAAUGAUGCGCAAAUGCGGAUAUCAAUGUAAAUCAGGGAGAAGGCAUGAUAUACCUUUAUAUCCGCAUUUGCGCAUCAUCUCU
((.(((((((((((((((((((((.((.(((.((((.(.......).)))).))).)).))))))))))))))))))))).)).
<------miRBase------> <------miRBase------>
*********************............................................................... length=21 depth=5
.......*********************........................................................ length=21 depth=2
........................................................*********************....... length=21 depth=1
............................................................*********************... length=21 depth=16
Thermodynamics stability
- MFE: Value of the Minimum Free Energy (computed by RNAeval)
- AMFE: Value of the adjusted MFE : MFE/(sequence length) x 100
- MFEI: Value of the minimum folding energy index (as defined here)
- Shuffles (option): Proportion of shuffled sequences whose MFE is lower than the MFE of the candidate miRNA precursor (see paragraph Compute thermodynamic stability). This value ranges between 0 and 1. The smaller it is, the more significant is the MFE. We report pre-miRNA stemloops for which the value is smaller than 0.01, which covers more than 89% of miRBase sequences. Otherwise, if the P-value is greater than 0.01, we say that it is non significant, and do not report any value.
HTML report for novel miRNAs
- Chromosome: Name of the chromosome
- Position: Start and end positions of the pre-miRNA, such as indicated in miRBase in 1-based notation (consistently with the GFF format). The length is indicated in parentheses.
- Strand: + (forward) or - (reverse).
- G+C content: Percentage of bases that are either guanine or cytosine.
- miRNA sequence: Sequence of the predicted miRNA.
- miRNA depth: Depth of the miRNA read (weight: miRNA depth divided by the number of possible alignments of this read in the genome.)
- Candidates with the same miRNA: List of other miRkwood predictions that involve the same miRNA sequence.
- Sequence (FASTA format): Link to download the sequence of the precursor.
- Stemloop structure: Link to download the secondary structure in dot-bracket format. The first line contains a FASTA-like header. The second line contains the nucleic acid sequence. The last line contains the set of associated pairings encoded by brackets and dots. A base pair between bases i and j is represented by a '(' at position i and a ')' at position j. Unpaired bases are represented by dots.
> 1:234009-234092,-, stemloop structure GAAAUGAUGCGCAAAUGCGGAUAUCAAUGUAAAUCAGGGAGAAGGCAUGAUAUACCUUUAUAUCCGCAUUUGCGCAUCAUCUCU ((.(((((((((((((((((((((.((.(((.((((.(.......).)))).))).)).))))))))))))))))))))).)).
- Optimal MFE secondary structure: If the stemloop structure is not the MFE structure, we also provide a link to download the MFE structure.
- Alternative candidates (dot-bracket format): This is the set of stemloop sequences that overlap the current prediction. The choice between several alternative overlapping candidate pre-miRNAs is made according to the best MFEI.
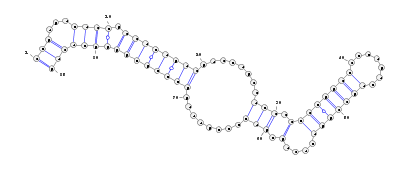
The stemloop structure of the miRNA precursor is also displayed with Varna.
Quality:
This score includes the reads ditribution score (as defined here) and extends it with some additional criteria. It ranges from 0 to 6.- MFEI < -0.8: This MFEI threshold covers 83% of miRBase pre-miRNAs, whereas it is observed in less than 13% of pseudo hairpins.
- Criteria number of reads: The locus has either at least 10 reads mapping to each arm, or at least 100 reads mapping in total.
- Existence of a miRNA: It is the sequence of the most common read, if the frequency is at least 33%.
- The miRNA is validated by miRdup: This measures the stability of the miRNA/miRNA* duplex (more information),
- Criteria presence of the miRNA:miRNA* duplex: There is at least one read in the window [-5,+5] on the strand of the miRNA*.
- Criteria precision of the precursor processing: At least 75% of reads start in a window [-3,+3] centered around the start position of the miRNA, or [-5,+5] on the opposite arm of the stemloop.
Reads
- Number of reads: The total number of reads included in the locus.
- Reads length distribution: This plot gives the sequence length distribution for all reads in the precursor locus.
- Reads cloud: This is a visual representation of all reads included in the locus.
Each ********** string is a unique read. Its length and its depth (the number of occurrences of this read in the total set of reads) are reported at the end of the dotted line. Each <------miRBase------> string indicates the existence of an alignmnent with a sequence from miRBase. More information on this alignment is provided in the sequel of the report, in Section Conservation of the mature miRNA.
Locus : 1:234009-234092
Strand : -
GAAAUGAUGCGCAAAUGCGGAUAUCAAUGUAAAUCAGGGAGAAGGCAUGAUAUACCUUUAUAUCCGCAUUUGCGCAUCAUCUCU
((.(((((((((((((((((((((.((.(((.((((.(.......).)))).))).)).))))))))))))))))))))).)).
<------miRBase------> <------miRBase------>
*********************............................................................... length=21 depth=5
.......*********************........................................................ length=21 depth=2
........................................................*********************....... length=21 depth=1
............................................................*********************... length=21 depth=16
Thermodynamics stability
- MFE: Value of the Minimum Free Energy (computed by RNAeval)
- AMFE: Value of the adjusted MFE : MFE/(sequence length) x 100
- MFEI: Value of the minimum folding energy index (as defined here)
- Shuffles (option): Proportion of shuffled sequences whose MFE is lower than the MFE of the candidate miRNA precursor (see paragraph Compute thermodynamic stability). This value ranges between 0 and 1. The smaller it is, the more significant is the MFE. We report pre-miRNA stemloops for which the value is smaller than 0.01, which covers more than 89% of miRBase sequences. Otherwise, if the P-value is greater than 0.01, we say that it is non significant, and do not report any value.
Conserved mature miRNA
All alignments with miRBase are reported and gathered according to their positions.
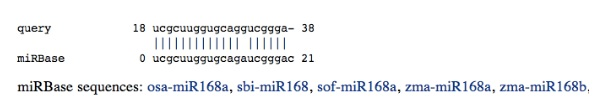
query is the user sequence, and miRBase designates the mature miRNA found in miRBase. It is possible to access the corresponding miRBase entry by clicking on the link under the alignment. Finally, we provide an ASCII representation of the putative miRNA within the stemloop precursor.