Table of contents
- Iedera overview
- Run in-browser
- Download
- Applications
- Subset seeds for AB SOLiD® read mapping
- Subset seeds for protein alignment
- Spaced seed design on profile HMMs for precise read mapping
- Transition seeds and new human/mouse alignments
- Spaced seed coverage and SVM string-kernels / k-mer distances
- Spaced seed dominance and hit integration / dirac / heaviside models
- Minimally-overlapping words seeds
- Usage
Iedera overview
Iedera is a program to select and design subset seed and vectorized subset seed patterns. Spaced seeds (see the quick introduction) and transition constrained spaced seeds can be perfectly represented in the subset seed model. Moreover, BLASTP-like seeds, and more generally Vector seeds (seeds with cumulative score constraint) can also be represented by vectorized subset seeds (which is a subset seed with an additional score constraint).
Iedera is applied to both lossy and lossless seed design. It is already used to design spaced seeds templates for read mappers (see below), to design subset seed templates for protein sequences (see below) or nucleic sequences (see below). Recent applications also include sub profile-HMM read mapping problem (see below), alignment-free distances and SVM string kernels with a new coverage criterion (see below), parameter-free spaced seed design (see below), and more recently minimally-overlapping words (see below) which are a better sampling alternative to minimizers.
This tool is experimental and is provided for research purposes only !! It natively supports two probabilistic models to describe the alignment sequence distribution (Bernoulli and Markov) and other models (as HMM) can be represented by probabilistic automata (see below).
The first version of Iedera was developed by L. Noe, G. Kucherov, and M. A. Roytberg. Since ~ 2010, Iedera is developed and maintained by L. Noe of the Bonsai team.
Main features of Iedera are: 
- the ability to choose a probabilistic lossy framework (default behavior) or a lossless framework to design seeds,
- the ability to set seed and alignment alphabets, and define their compatibility with a boolean matching matrix,
- the ability to generate seeds randomly or enumeratively, with constraints on size, weight, number of hits, placement, excluded seeds, and even letter composition (signature), or to simply check a given set of seeds by specified in command line,
- the ability to switch from subset seed to vectorized subset seed model.
If you use this program in your work, please cite one of these papers:
[1] G. Kucherov, L. Noe, M. Roytberg, Subset seed automaton, Proceedings of the 12th International Conference on Implementation and Application of Automata (CIAA 2007), LNCS 4783:180-191 , 2007.
[2] G. Kucherov, L. Noe, M. Roytberg, A unifying framework for seed sensitivity and its application to subset seeds, Journal of Bioinformatics and Computational Biology, 4(2):553-569, 2006.
If you still need it, the old precursor named hedera is still there.
Your are welcome! We always appreciate to share new ideas and problems...
top^Run in-browser
🆕 We now provide a webassemby console to run the iedera command line in-browser.
top^Download
- Source Code: v1.07 α3 [build jul 1, 2021] [tar.gz, zip]
- Binaries: v1.07 α3 [build jul 1, 2021] [win64-x86_64, mac-os-x]
- Github : <https://github.com/laurentnoe/iedera>
- Older versions can be found here [dir]
Applications
Subset seeds for Applied Biosystems SOLiD® read mapping
The method has been extended and applied to design seeds on HTS reads, taking into account both the DNA variability and also reading errors [3,4]. We have an additional webpage that describes the technical details to design seeds for the SOLiD® scheme + a read mapping tool.
- Associated paper is published here, preprint is avalaible here, and final journal version is here,
- Supplementary results are given here and here,
- Software used in the last experimental part (SOLiD read-mapping) is given here.
[3] L. Noe, M. Girdea, G. Kucherov, Seed design framework for mapping SOLiD reads, Proceedings of the 14th Annual International Conference on Research in Computational Molecular Biology (RECOMB 2010), LNCS 6044:384-396, 2010.
[4] L. Noe, M. Girdea, G. Kucherov, Designing Efficient Spaced Seeds for SOLiD Read Mapping, Advances in Bioinformatics, Article ID 708501, 2010.
top^Subset seeds for protein alignment
Subset-seeds have been used to detect protein homologies. Details of the models used, together with experiments to design seeds and measure their sensitivity on real datasets are given in the paper On subset seeds for protein alignment [5] and Subset Seed Extension to Protein BLAST [6]. We have an additional webpage that describes the technical details to design seeds for proteins.
- Associated paper is published here, preprint is avalaible here,
- Supplementary results are given here.
Moreover, a new software named plastp has been developped by Van Hoa Nguyen and Dominique Lavenier of the INRIA Symbiose team. It is similar in use to blastp (by now, it provides plastp, tplastn, plastx and tplastx) but is highly parallel (both efficient SIMD and Multithreading) and uses subset seed patterns: I thus give this script to transform seeds alphabets and seed patterns obtained with iedera into readable seeds for plastp.
- Plastp paper and webpage,
- Script to convert seeds generated with iedera to seeds used by plastp is given here.
[5] M. Roytberg, A. Gambin, L. Noe, S. Lasota, E. Furletova, E. Szczurek, G. Kucherov, On Subset Seeds for Protein Alignment, IEEE/ACM Transactions on Computational Biology and Bioinformatics, 6(3):483-494, 2009.
[6] A. Gambin, S. Lasota, M. Startek, M. Sykulski, L. Noe, G. Kucherov, Subset Seed Extension to Protein BLAST, International Conference on Bioinformatics Models, Methods and Algorithms (Bioinformatics), 149-158, 2011.
top^Spaced seed design on profile HMMs for precise HTS read-mapping
[ongoing] (iedera v1.06 α ≥ 5) Spaced seeds have been also designed to comply with the restricted alignment length that a read produces, when aligned against a profile-HMM, itself combined with the variable conservation level along the profile-HMM : seeds can thus be designed / position-designed on the full profile-HMM, by taking into consideration this conservation level along all the possible read alignments (windows) on this profile-HMM. We have a draft paper and an additional webpage that describes the experimental details.
top^Transition seeds and new human/mouse alignments
How better transition seeds may help finding new significant alignments when the transition/transversion rate is high? Last software (with Iedera seed design) may help [7] :
[7] M. C. Frith, L. Noe Improved search heuristics find 20 000 new alignments between human and mouse genomes, Nucleic Acids Research, 42(7):e59. 2014
top^Spaced seed coverage and SVM string-kernels and k-mer distances applications
(iedera v1.06 α ≥ 7) Spaced seeds have been used recently in alignment-free distances and SVM string-kernels. We have adapted a new coverage criterion (defined as the number of matches covered by at least one maching symbol from one seed hit) to comply with these new problems. Details of the models used, together with experiments to measure seeds are given in the paper A Coverage Criterion for Spaced Seeds and Its Applications to Support Vector Machine String Kernels and k-Mer Distances [8]. We have an additional webpage that describes the technical details.
- Associated paper is published here, preprint is avalaible here,
- Supplementary results are given here and here.
[8] L. Noe, D. E.K. Martin A Coverage Criterion for Spaced Seeds and Its Applications to Support Vector Machine String Kernels and k-Mer Distances, Journal of Computational Biology, 21(12). 2014
top^Spaced seed dominance and applications to hit integration, dirac or heaviside models / lossless seeds
(iedera v1.06 α ≥ 9) Spaced seeds have been selected by Mak & Benson 2009 with a new spaced seed dominance criterion on the Bernoulli model; we extend their work on the Hit integration model, and propose two discrete Dirac and Heaviside models that can carry lossless seeds in the same framework. You will find the whole description in the paper Best hits of 11110110111: model-free selection and parameter-free sensitivity calculation of spaced seeds [9], you can also get the full experimental results on this additional webpage that provides the scripts, some technical details, and many plots (pdf or html) where you can pick good seed patterns according to your needs.
- Associated paper is published here, preprint is avalaible here,
- Extra plots are given in pdf or html,
- Supplementary results are given here and here.
[9] L. Noe, Best hits of 11110110111: model-free selection and parameter-free sensitivity calculation of spaced seeds, Algorithms for Molecular Biology, 12(1). 2017
top^Minimally-overlapping (spaced-like) words for sequence similarity search
(iedera 1.07 hfkn_check / iupac branch) Minimally-overlapping words have been shown to be better - more sensitive - than minimizers for selecting conserved k-mers, in Frith & all 2020. We try to further optimize the main result at the end of the paper with a spaced-like pattern, and with a quite good success.
- Associated paper is published here, preprint is avalaible here,
- Main experimentals scripts and extra tools from Martin C. Frith are provided here.
[10] M.C. Frith, L. Noe, G. Kucherov, Minimally-overlapping words for sequence similarity search, Bioinformatics, 36(22-23):5344-5350, 2020.
top^Usage
Quick introduction
If you simply need to design spaced seed templates (with the commonly used default parameters), try this first:
(1) to design one spaced seed pattern of weight 9, span (seed length) from 9 to 15, type:
./iedera -spaced -w 9,9 -s 9,15 (or ./iedera.exe -spaced ... )
you will obtain the following result, where the seed pattern is given by a set of '#' and '-':
##-##-#-#---### 0.999996 0.729156 0.270844
first column gives second third is last is distance
the seed gives sensitivity to (1,1) coordinate
selec- in sel/sen.
tivity
(2) to design one spaced seed template of weight 11, span from 11 to 18, type:
./iedera -spaced -w 11,11 -s 11,18
(3) to design 2 complementary spaced seed patterns of weight 11, span from 11 to 20, randomly
drawn 10000 times with an additional hill-climbing heuristic, type:
./iedera -spaced -w 11,11 -s 11,20 -n 2 -r 10000 -k
(4) a good change can be to reset the default alignment length to 40 (on previous examples, it
was set to 64), type:
./iedera -spaced -w 11,11 -s 11,18 -l 40
and see the difference: smaller span of the seed been selected and also lower sensitivity on
shorter alignments compared with (2).
(5) to design "lossless" seeds (with up to 2 mismatches for an alignment of length of 25), type :
./iedera -spaced -L 1,0 -X 2 -l 25 -w 12,12 -s 18,20 -r 10000 -k
and see the explanation below.
More details:
To understand subset seeds, the first objects that need to be described are the alignment alphabet A and the seed alphabet B built upon A.
Let start with A: alignments can be represented by a succession of mismatches or matches (at first sight), thus with a simple binary definition for A : A = {'0','1'}. Each seed letter b of B is defined as a subset of the alignment alphabet A ; e.g. if A = {'0','1'} then there are four choices for b: b is either the empty set {}, either one of partial sets {'0'} or {'1'}, or the full set {'0','1'}.
A seed letter b "detects" a if the subset describing b contains a. E.g. if we consider that A = {'0','1'} is defined by '0' for mismatch, '1' for match, then the seed letter '-' defined by the subset {'0','1'} is commonly called jocker since it "detects" any letter of A. The seed letter '#' defined by the subset {'1'} is called a must match letter since it only "detects" the match symbol '1' of A. Note that the two other possible seed letters (empty set {} and "must mismatch" {'0'}) are by no way useful to detect interesting alignments.
A seed π defined as a word on B (π = b1b2...bs) "detects" the alignment defined as a word on A (a1a2...an) at a given position p (0 ≤ p ≤ n - s) iif for all i (1 ≤ i ≤ s) bi contains ai+p.
E.g if we consider that A = {'0','1'}, B = {'-','#'} where '-' is defined by the subset {'0','1'} and '#' is defined by the subset {'1'}, then the seed π='##-#' detect the alignment '101101111' at the 3th and 6th positions (thus for p = 2 and p = 5):
101101111 ||||||| ..##-#||| .....##-#
Finally, for a given seed π to "detect" an alignment, we have to find at least one position p satisfying the previous condition. iedera does this checking in parallel on the full set of "interesting" alignments using automata theory (see paper [2] for more details).
Now, we can show into more technical details how to set the alignment alphabet, the seed alphabet, and their compatibity matrix in iedera:
(1) Default alignment alphabet A and seed alphabet B are of size 3. Note that you can modify both
sizes using the -A and -B parameters. Alignment and seed alphabet letters are represented
(by default) with the symbols "0..9A..Za..z": thus, an alphabet of size 3 is composed of the
symbols '0','1','2'.
As alignments and seeds have the same set of symbols by default, it is a good idea to
override B symbols with the following parameter:
-BSymbols '-@#'
(2) The subsets are given by a boolean matrix (subset matching matrix) of B lines and A columns.
The default boolean subset seed Matching matrix is defined as an upper triangular:
{{1,1,1},
{0,1,1},
{0,0,1}}
each seed letter is represented by one line inside this boolean matrix:
- seed letter '0' or '-' (1st line) is called joker since it accepts any letter of A: {1,1,1}
- seed letter '1' or '@' (2nd line) accepts the 2nd & 3rd letter of A but not the 1st: {0,1,1}
- seed letter '2' or '#' (3rd line) is usually a must match letter, since it only accepts the
last letter '2' of A which usually represents a match.
You can modify this binary matrix using the -M option:
./iedera -A 3 -B 4 -M \{\{1,1,1\},\{0,1,1\},\{1,0,1\},\{0,0,1\}\} -BSymbols '-@x#'
in this example, we have defined 4 seed letters on alignment alphabet of size 3:
'-'={'0','1','2'}, '@'={'1','2'}, 'x'={'0','2'} and '#'={'2'}
Once the two alphabets are defined (and the subset matrix possibly set), we need to define probabilistic distributions for the alignment alphabet: two of them are needed to describe the foreground distribution (for true "target" alignments) and background distribution (random "bad" alignments). We choose here to use the simplest Bernoulli model.
(3) Each alignment letter of A has a foreground and background probability to appear,
according to models (Bernoulli or Markov), look at the -f and -b commands.
(a) A commonly used parameter for spaced seeds is "-f 0.3,0.7 -b 0.75,0.25" that gives, for the
alignment alphabet A, a foreground probability of 0.7 for the match symbol (respectively of
0.3 for mismatch symbol), and a background probability of 0.25 for the match symbol
(respectively of 0.75 for mismatch symbol).
(b) A classical example to design spaced seeds of weight 11, span 12 to 18, on Bernoulli
alignments of length 64, using previous probabilities:
./iedera -A 2 -B 2 -l 64 -w 11,11 -s 11,18 -f .3,.7 -b .75,.25
111010010100110111 1 0.467122 0.532878
first column gives second third is last is distance
the seed gives sensitivity to (1,1) coordinate
selec- in sel/sen.
tivity
It is possible to use the "-spaced" parameter that represents shortcut for
"-A 2 -B 2 -f .30,.70 -b 0.75,0.25 -BSymbols '-#' -l 64 -w 9,9 -s 9,15".
You only have, when needed, to override weight and span (e.g. "-spaced -w 11,11 -s 11,19").
(c) Another example to design transition-constrained seeds of weight 11, span 12 to 18:
./iedera -A 3 -B 3 -l 64 -w 11,11 -s 12,18 -f .15,.15,.70 -b .50,.25,.25 -r 10000
221022002120102122 1 0.476753 0.523247
here we focus on probabilities:
-f .15,.15,.70 gives the probability to have a transversion, a transition, or a match
in a target ("good") foreground alignment,
-b .50,.25,.25 does exactly the same, but for a random ("bad") background alignment.
It is also possible to use the "-transitive" parameter that represents shortcut for
"-A 3 -B 3 -f 0.15,0.15,7 -b 0.5,0.25,0.25 -BSymbols '-@#' -l 64 -w 9,9 -s 10,15 -i 0,2,8".
You have, when needed to override weight, span, and signature that
represents the number of symbols '(-)@#' allowed in a seed
(e.g. "-transitive -w 11,11 -s 12,19 -i 0,2,10").
(d) A third example with 2 seeds of weight 12, span 12 to 19:
./iedera -A 3 -B 3 -l 64 -w 12,12 -s 12,19 -f .15,.15,.70 -b .50,.25,.25 -r 50000 -k -n 2
2220211022120020122,2121022020012201222 1 0.500499 0.499501
Here, we cannot use full enumeration of all the pair of seeds (would take decades), so we use
the "-r 50000 -k" parameter to randomly generate pair of seeds with a hill-climbing heuristic
Finally, it is possible to design seeds by taking into account the fact that they are lossless according to a given criterion: in this case, sensitivity (e.g "0.9790") is replaced by "[lossless]" when the seed is lossless. Note that a seed with sensitivity equal to 1.0 does not mean that it is lossless (although not far from being perfect ... there are still a few alignments that escape)
(4) About the design of lossless spaced seeds:
(a) we want to find a seed of weight 12 that matches all alignments of length 25 with
at most 2 mismatches: we thus fix a penalty of 1 for a mismatch and 0 for a match
(-L 1,0) and set a max penalty of 2 (-X 2)
./iedera -spaced -s 12,19 -w 12,12 -l 25 -L 1,0 -X 2
#-###--#-###--#-### 1 [lossless] 5.96046e-08
(b) if we want to find several (eg. 2) seeds, full enumeration is unusable, so we hope
to be lucky with these parameters after one full night ...
./iedera -spaced -l 25 -L 1,0 -X 2 -n 2 -s 20,21 -w 14,14 -r 100..some.zeros..00 -k
####-#-##--####-#-##,#-##--####-#-##--#### 1 [lossless] 7.45058e-09
Command line parameters
iedera [options]
- -h display this Help screen
- -v display program Version
Seed Model:
Data
- -A <int> : align alphabet size 'A' (default 3)
- -B <int> : seed alphabet size 'B' (default 3)
- -M {{<bool>}} table : subset seed Matching matrix
(default {{1,1,1},{0,1,1},{0,0,1}} )- note : parameter is a C-like matrix of 'A'x'B' cols x rows
- example : -M "{{1,0,1},{0,1,1}}" (for A=3 and B=2)
- -MF <filename> : gives the matrix inside a file (when it is too large for command line)
SubsetSeed/VectorizedSubsetSeed
- -V : select the Vectorized subset seed model (default 0)
- -S {{<int>}} table : vectorized subset seed Scoring table
- -SF <filename> : gives the table inside a file (when it is too large for command line)
- -T <int> : vectorized subset seed scoring Threehold
SubsetSeed/LosslessSubsetSeed
- -L <int>,<int>,... : set the Lossless alignment alphabet costs (and select the lossless model)
- -X <int> : set the alignment maX cost to be considered in the alignment set
Alignments:
- -b <double>,<double>,...: fix the Bernoulli/Markov background distribution (size |'A'|^k)
- -f <double>,<double>,...: fix the Bernoulli/Markov foreground distribution (size |'A'|^k)
- -fF <filename> : fix the foreground model (as a probabilistic NFA)
for more details, see https://bioinfo.univ-lille.fr/yass/iedera.php#fFFormat - -l <int> : fix the alignment length (default = 16)
- -u <int>,<int>,... : set the homogeneous alignment scoring system (default = disabled)
Seed Enumeration:
- -s <int>,<int> : seed span (default min=1,max=5)
- -n <int> : number of seeds (default 1)
- -r <int> : random enumeration (default 0 is complete enumeration)
- -k : hill climbing heuristic (check all permutations of 2 elements inside one seed)
- -a <double> : hill climbing heuristic frequency (good seeds are more likely to be optimized)
- -w <double>,<double> : minimal/maximal weight (default min=-1e+32,max=1e+32)
- symbol # (if defined othewise the max of all symbols of all symbols) is of weight 1.0
- symbol _ is 0.0
- symbol e is given by log_#(e matches background distribution)
- -i <int>,<int>,... : signature id (number of '0','1',...'B-1' elements inside a subset seed)
- -j <double> : difference of weight (given as a ratio) allowed between consecutive seeds (value is between 0 and 1, default 0)
- -x : only symetric seeds are selected [beta]
- -y <int> : consider sensitivity when y hits are required (multihit sensitivity) [beta]
- -c <int>,<int>,... : consider sensitivity when each seed is indexed on 1/c of its positions
- note : the position is enumerated or choosen randomly depending on -r parameter
- example : "##-#:1/5" means that the seed "##-#" will be placed at 1st,6th,11th... positions
- -q <int>,<int>,... : consider sensitivity when each seed is indexed on q/c of its positions
- note : parameter -c must be specified before, positions are enumerate or randomly drawn (-r)
- example : "##-#:2,5/5" means that the seed "##-#" will be placed at 2nd,5th,7th,10th... positions
- -d : disable Hopcroft minimization (default is enabled)
- -m <seeds> : check the given seed selectivity/sensitivity (possibility to add cycle content : see -c -q examples)
- -mx <seeds> : generate alignments that exclude the given seed pattern
Miscellaneous:
- -BSymbols '<char>' : chain of seed alphabet symbols (default is '01..9AB..Zab..z')
- -e <filename> : first select an initial "pareto set" file before optimization
- -o <filename> : write the "pareto set" to this file after optimization
Nucleic spaced seeds: (shortcuts that simulates hedera parameters)
- -transitive : "-A 3 -B 3 -f 0.15,0.15,7 -b 0.5,0.25,0.25 -BSymbols '-@#' -l 64 -w 9,9 -s 10,15 -i 0,2,8"
- note : when you change the weight (-w), dont forget to change the signature (-i)
- -spaced : "-A 2 -B 2 -f 0.3,0.7 -b 0.75,0.25 -BSymbols '-#' -l 64 -w 9,9 -s 9,15"
Probabilistic automaton file format
A small but detailled example:
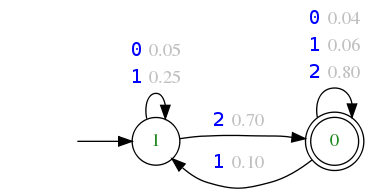
First line gives the number of states (2 in the next example). Then each state is described (its current state number is given and a final flag is set). For each state, next lines describe a list of transitions that start from this state: for each letter of the alignment alphabet (here 0, 1 or 2), the number of transitions outgoing from the current state on the given letter is provided, followed on each new line by a single transition (more precisely, its destination state and its probability). Finally, note that the initial state is by convention always the state 1.
2 → number of states 0 1 → Current state : current state number + final state flag 0 1 → alignment alphabet letter + number of transitions outgoing from current state on this letter 0 0.04 → Current transition : destination state number + probability to follow this transition 1 2 0 0.06 1 0.10 2 1 0 0.80 1 0 0 1 1 0.05 1 1 1 0.25 2 1 0 0.70
The previous description is equivalent to this probabilistic automaton: